

Numerose sono le patologie correlate al metabolismo dei carboidrati: esse sono causate da alterazione nella sintesi, scissione e smaltimento dei carboidrati. Inoltre molte di queste patologie sono ereditarie e tra queste ci sono le glicogenosi, la galattosemia, la pentosuria, la fruttosuria e le intollerante a lattosio e saccarosio.
In questo articolo verranno illustrate in particolare le glicogenosi, andando a illustrare quali sono le mutazioni e gli enzimi implicati.
Indice dell'articolo
Cosa sono le glicogenosi?
Le glicogenosi sono patologie dovute ad alterazioni metaboliche della sintesi, smaltimento e utilizzo del glicogeno, che, pertanto, viene accumulato all’interno della cellula.
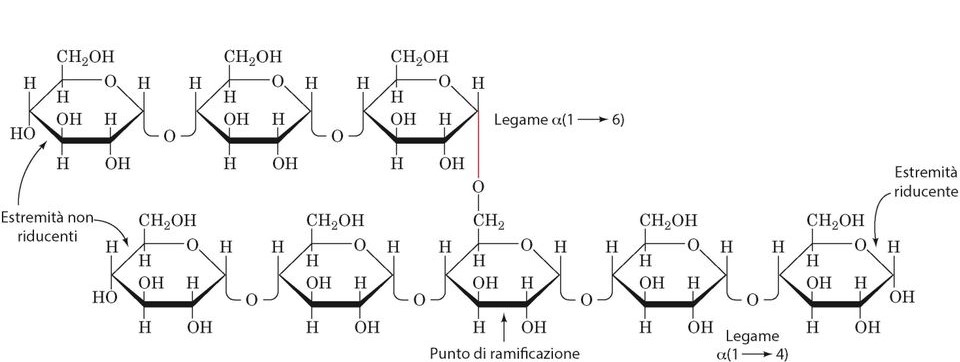
Il glicogeno è un omopolimero ramificato del glucosio con legami alfa 1,4 glicosidico e alfa 1,6 glicosidico presente ogni 10 residui. Dunque, si tratta di una molecola con una struttura compatta utilizzata da molti tessuti come fonte di energia rapida.
Le glicogenosi sono causate da accumulo di glicogeno perché la cellula non riesce ad utilizzarlo. Infatti le glicogenosi sono causate da difetti enzimatici dovute quasi sempre a mutazioni autosomiche recessive.
Gli organi maggiormente colpiti sono fegato, muscoli e reni. Essi infatti sono quelli più attivi nel metabolismo del glicogeno e, pertanto, risultano maggiormente danneggiati dall’anomalo accumulo della molecola.
Incidenza delle glicogenosi nella popolazione
La glicogenosi ha un’incidenza nella popolazione di 1/20.000 – 25.000 nati (a seconda delle etnie). Tuttavia questo valore è sottostimato in quanto alcune forme di glicogenosi non sono diagnosticate. Le più frequenti nella popolazione sono le glicogenosi I e III tra le forme epatiche, e la glicogenosi II tra le forme muscolari.
Classificazione delle glicogenosi
Ci sono numerose forme di glicogenosi che vengono riunite in tre gruppi:
- epatiche (colpiscono maggiormente il fegato);
- muscolari (colpiscono prevalentemente l’apparato muscolare);
- miste.
Forme epatiche di glicogenosi
Tipo I
La glicogenosi tipo I è dovuta ad un difetto dell’enzima epatico glucosio-6-fosfatasi la cui funzione è quella di rilasciare glucosio libero a partire dal glicogeno; in questo modo il fegato può liberarlo in circolo per regolare la glicemia.
Un danno di questo enzima provoca ipoglicemia a digiuno. Nel tempo però il soggetto affetto da glicogenosi I si abitua a tollerare l’ipoglicemia, pertanto può essere asintomatico anche in presenza di bassi valori di glucosio ematico. Tuttavia, se i periodi di digiuno sono lunghi i tessuti periferici hanno poco glucosio e bisogna provvedere ad una dieta adeguata.
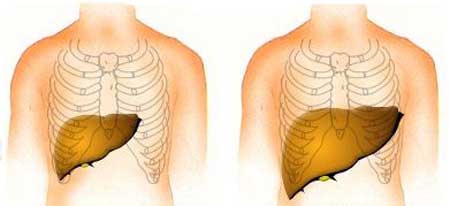
Il glicogeno non utilizzato inoltre si accumula nel fegato e anche nei reni provocando epatomegalia e nefromegalia. Altri sintomi clinici possono essere dovuti a difetti di crescita.
Diagnosi della glicogenosi di tipo I

La diagnosi è perlopiù clinica e viene effettuata in funzione della sintomatologia e dei test di laboratorio. Si effettua il dosaggio della glicemia a digiuno; inoltre si valuta l’acidosi lattica e l’iperuricemia. Invece la diagnosi molecolare si basa sul sequenziamento del gene che codifica per l’enzima glucosio-6-fosfatasi e del gene SLC37A4, che codifica per il trasportatore del glucosio-6-fosfato.
Tipo III
Questa forma di glicogenosi è dovuta ad un difetto dell’enzima deramificante o AGL (attività alfa 1- 6 glicosidasica). Se il glicogeno non può essere deramificato, si accumula in forma anomala e viene prodotto glicogeno igroscopico, una forma che occupa molto più spazio nella cellula. Inoltre questo deficit può essere presente sia a livello epatico (tipo A) che muscolare (tipo B).
Tipo IV
È dovuta alla mutazione dell’enzima ramificante, chiamato glicosil 4 – 6 transferasi. Il glicogeno privo di catene laterali risulta igroscopico. Ciò causa un aumento delle dimensioni cellulari, provocando epatomegalia e progressivo danno che si manifesta come cirrosi epatica. Pertanto le glicogenosi di tipo IIIa e IV risultano forme piuttosto simili.
Tipo VI
Essa è dovuta ad un difetto dell’enzima fosforilasi epatica. Questa è una variante asintomatica od oligosintomatica, per cui nella maggior parte dei casi non è nemmeno diagnosticata.
Tipo IX
Questa forma è causata da un difetto della fosforilasi-chinasi e ha una trasmissione X-linked: ciò significa che i pazienti sono praticamente solo maschi.
I pazienti durante l’infanzia hanno addome globoso, presentano ritardo della crescita, ipercolesterolemia, aumento dei trigliceridi, transaminasi, acetone a digiuno e modesta ipoglicemia. Tuttavia queste alterazioni scompaiono con l’età, infatti gli adulti sono considerati asintomatici. Perciò si tratta di una glicogenosi che presenta un decorso favorevole e oligosintomatico.
Forme muscolari di glicogenosi
Tipo II
È anche chiamata tesaurismosi, termine con il quale sono indicati processi morbosi che coinvolgono le malattie lisosomiali. Infatti colpisce l’enzima maltasi acida dei lisosomi, il cui gene è localizzato sul cromosoma 17.
Inoltre, anche se è classificata come forma muscolare, la glicogenosi di tipo II è considerata una patologia sistemica. Pertanto tutte le cellule soffrono di tale disfunzione. Tuttavia il muscolo scheletrico è l’organo più compromesso perché coinvolto nell’immagazzinamento del glicogeno.
La glicogenosi di tipo II è considerata molto grave, in quanto la sua sintomatologia peggiora con l’avanzare dell’età. Ci sono numerose varianti, ma la forma la forma più grave è chiamata malattia di Pompe. I soggetti affetti da questa forma nei primi anni di vita sono caratterizzati da ipotonia muscolare e cardiomegalia; infine, la malattia di Pompe causa la morte già nei primissimi anni di vita
Diagnosi della glicogenosi di tipo II
Essendoci analogie con la distrofia muscolare di Duchenne, è necessaria una accurata diagnosi differenziale. Infatti a livello molecolare bisogna ricercare le mutazioni dei geni implicati in entrambe le malattie Inoltre può essere necessaria la biopsia muscolare e il dosaggio dell’attività enzimatica.
Tipo V
Chiamata anche malattia di McArdle, è dovuta ad un’isoforma muscolare della glicogeno fosforilasi, con conseguente blocco della glicogenolisi a livello muscolare, senza compromissione dei reni e del fegato. In questi soggetti la mancata glicogenolisi comporta accumulo di glicogeno nel muscolo. Pertanto in caso di sforzo si hanno crampi, dolore muscolare e aumento della mioglobinuria (le urine sono scure).
La glicogenosi di tipo V è caratterizzata da una sintomatologia lieve, dunque la diagnosi è solitamente tardiva. Il soggetto che ne è affetto avverte i primi sintomi durante l’età adulta e dopo sforzi di elevata intensità e di breve durata. Tuttavia la gravità dei sintomi dipende dalle mutazioni e dalle varianti alleliche più o meno gravi.
Tipo VII
Questa variante è dovuta ad un’alterazione della fosfofruttochinasi muscolare, il cui gene è situato sul cromosoma 1. Tale enzima favorisce l’ingresso di glucosio durante la glicolisi. Pertanto, se l’enzima non funziona correttamente, il glucosio resta libero e si accumula come glicogeno. I sintomi sono simili a quelli della malattia di McArdle. Tuttavia ci sono altri sintomi come nausea e vomito dopo un pasto ricco di carboidrati.
Terapia delle glicogenosi
Negli ultimi anni sono state sviluppate alcune terapie per il trattamento delle glicogenosi più gravi. Quella più studiata è senza dubbio la glicogenosi di tipo II (malattia di Pompe) a causa dell’estrema gravità della patologia.
Le strategie per trattarla si basano su principalmente due: la Terapia Enzimatica Sostitutiva (ERT) e la terapia genica.
- la Terapia Enzimatica Sostitutiva (quella attualmente più utilizzata) consiste nel somministrare una versione artificiale dell’enzima che manca ai pazienti con malattia di Pompe, ovvero la alfa-glucosidasi acida. In questo modo si riesce a recuperare la forza muscolare fin dai primi anni di vita.
- la terapia genica invece cerca di inserire il gene sano nel DNA del paziente sfruttando dei virus geneticamente modificati. Infatti è stato condotto un importante studio clinico dai ricercatori dell’Università della Florida su un tipo di terapia genica denominata AAV1-CMV-GAA. Questa ha dato risultati incoraggianti mostrando un miglioramento della funzionalità respiratoria dei pazienti, soprattutto di quelli più gravi.
Roberta Miele
Webgrafia
https://www.ncbi.nlm.nih.gov/books/NBK1312/
https://www.ncbi.nlm.nih.gov/books/NBK26372/
https://www.ncbi.nlm.nih.gov/books/NBK1344/
https://www.ncbi.nlm.nih.gov/books/NBK5941/